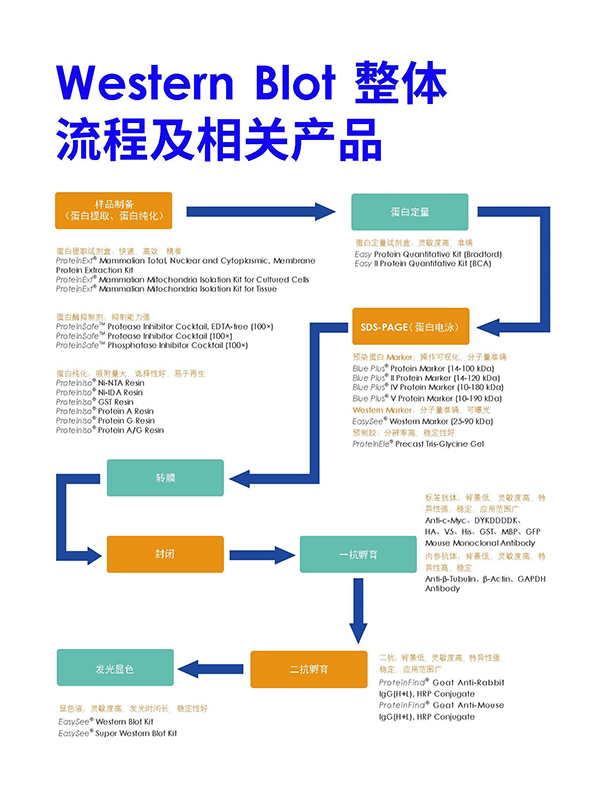
1. 转膜失败
观察预染Marker,没有Marker条带(一条都看不到)可能的原因:
①考虑电极是否插反,若是,需要重新进行电泳及转膜;
②使用的是PVDF膜,要考虑转膜前是否使用无水甲醇进行了活化。
转膜不充分(小条带在膜上,大条带在胶上)的可能原因有:
①转膜时间过短,需适当延长转膜时间;
②出现不均匀转膜,表示膜没有经过充分的浸润平衡,需要延长膜在转膜缓冲液中的平衡时间。
过转(大条带在膜上,小条带消失)的可能原因:
①转膜电压太高或时间过长,减低电压,减少转膜时间;
②膜的孔径不合适,要根据蛋白大小选择合适孔径的膜(0.2 μm或更小);
③蛋白与膜的结合不强,可以选择结合能力更强的PVDF膜
2. 没有信号或信号很弱
这一问题要分为两种情况:一种是上面提到的转膜失败,建议使用丽春红染色或预染Marker,来监测转膜状态。
另一种是无目的条带或条带很弱,可能原因有多种:
①蛋白上样量过低,需要增加上样量;
②抗体效价、浓度低,需要增加抗体浓度或延长孵育时间;
③抗体失效,这种常见于反复冻融导致的失效,需要更换或补加新的抗体;
④ECL显色液失效,ECL显色液需要现用现配;
⑤蛋白制备过程中降解或储存不当导致降解,重新制备样品;
⑥抗体浓度过高,导致底物迅速被消耗,在膜上可以看到淡黄色或浅褐色条带,需要对抗体进行一定程度的稀释。
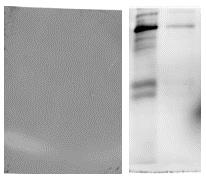
3. 背景过高

上面两张图片的背景很高,而且可以看出有非特异性条带,出现这种情况首先考虑是否为封闭不彻底,建议延长封闭时间(4℃晃动过夜)、增加封闭蛋白(如BSA、脱脂奶)的浓度、更换封闭液种类(将脱脂奶更换成BSA)等。其次考虑是否为洗膜不彻底,建议少量多次洗涤,一般条件为洗3次,每次洗涤10分钟,背景过高或有非特异性杂带时,可以考虑将洗膜缓冲液少加一些,洗膜次数增加成5次甚至更多,每次洗涤5分钟;也可以适当增加去垢剂的强度,如将Tween-20换成NP-40。另外,也可能由于抗体浓度过高或蛋白上样量过高导致,建议适当增加抗体的稀释倍数(主要为一抗),降低蛋白上样量。
4. 反影或空斑

如左图所示,反影或发现膜上的条带为黄色,说明蛋白上样量过高,或抗体浓度过高,需适当减少上样量或增加抗体稀释倍数。
右图中显色条带有白色未显影空斑,可能是膜未充分浸润平衡,用缓冲液或甲醇充分浸润膜;也可能是转膜时膜与胶之间有气泡,或显色后包保鲜膜时有气泡,需要仔细操作,赶走气泡,然后再进行下面的操作。
5. 微笑或皱眉条带
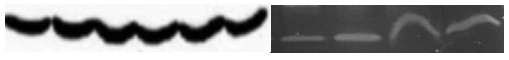
左图为微笑条带,可能的原因有:
①条带迁移过快或电泳温度过高,建议降低电压,缓慢电泳,如有需要可进行低温电泳;
②凝胶中间凝固不均匀(多见于厚胶),建议配胶后放置足够长的时间,待凝胶充分凝固后再进行电泳。
右图为皱眉条带,可能由凝胶与玻璃挡板底部的气泡导致,在上样之前可使用大量程移液枪反复吹打以排除气泡。
6. 条带扭曲

上图中的情况也比较常见,可能原因有两点:
①分离胶与浓缩胶界面不平,可以使用异丙醇或乙醇来进行封胶,之后需要用水进行润洗,另外做好分离胶后要注意立即封胶;
②蛋白溶解度不好或体系中盐离子浓度过高,上样前可以将蛋白进行离心,以去除析出颗粒的影响,另外需要优化蛋白提取方法,寻找更合适的裂解液。
7. 条带太宽或太粗

左图的条带太宽,可能是样品中盐离子浓度过高,需要调整裂解液的组成;也可能由于样品上样体积过大,电泳过程中样品向旁边泳道扩散,建议将所有泳道的上样体积调成一致。
右图的条带比较粗,可能是浓缩胶没有发挥作用,建议检测试剂的pH值,重新配制浓缩胶,另外可以适当增加浓缩胶的长度,尽量低电压进行电泳。
8. 斑点、纹理或拖尾
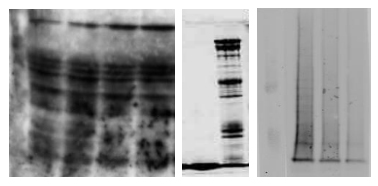
左图中有很多斑点,这个说明封闭液有杂质,在配制脱脂奶之后最好静置一段时间,使较大的颗粒沉淀下来。
中间的图片显示有垂直纹理,可能是样品中有不溶性颗粒,需要优化蛋白提取方法,寻找更合适的裂解液,上样前最好进行离心;还可能因为凝胶中有小气泡,在配胶时要注意观察并赶走气泡。
右图显示为拖尾,可能是蛋白样品溶解度不好,需要优化蛋白提取方法,寻找合适的裂解液,或者加样前进行离心;可能因为电泳缓冲液重复使用次数过多,需要更换新鲜的电泳缓冲液;也可能是因为分离胶浓度过大,需要降低凝胶浓度。
9. 其它问题
显色后目的蛋白分子量同预期不一致,可能原因有:
①蛋白翻译后修饰的影响,比如糖基化、磷酸化、羟基化修饰,都会影响到蛋白的实际大小。
②蛋白由特殊氨基酸组成,含有较多碱性氨基酸会使迁移率降低,如溶菌酶;含有较多酸性氨基酸会使迁移率增加。
③蛋白上样量太多,有可能导致条带下移,需要减少上样量判断准确位置。
电泳染色后加样孔底部或浓缩胶与分离胶界面处有条带,这种情况多发生于大分子量蛋白分析中,由于还原剂在加热的过程中被氧化而失去活性,致使原来被解离的蛋白质分子重新折叠或亚基重新聚合形成大分子,不能进入分离胶。解决办法有:加热煮沸样品后,添加适量的DTT或β-ME,以补充不足的还原剂;也可以在煮样时添加适量EDTA,以阻止还原剂的氧化。
当需要使用不同抗体对同一张膜进行杂交时,需要做Strip处理。Stripping Buffer的配方:100 mM 2-ME+2% SDS+62.5 mM Tris (pH 6.7),建议现用现配。将膜放置于Stripping Buffer中,55℃孵育30 min。之后使用TBS或TBST进行洗涤,并重新进行封闭、抗体孵育、显色等步骤。
整个 Western Blot 技术流程及可能出现的问题解析就给大家介绍到这里,那么如何保证WB的成功呢?
Western Blot 实验注意事项
①首先蛋白制备是关键,要注意冰上裂解,全程在低温下进行。
②凝胶的配制不可小觑,凝胶时间要足够长,以保证充分交联,也可放置于4℃进行交联。
③蛋白上样前,建议使用电泳缓冲液清洗梳孔,以去除气泡和碎胶,防止出现条带扭曲现象。
④上样时建议各泳道中蛋白样品的上样体积保持一致,可以使用1×Loading Buffer将上样体积调成一致。
⑤如果条件允许,可以低电压跑浓缩胶,再调高电压跑分离胶。封闭和洗膜一定要充分,以减少非特异性条带及背景。
⑥一抗和二抗的浓度要进行优化,浓度过高或过低都无法得到好的条带。科学的设置阳性对照和阴性对照,以监测整个过程。
要确保实验成功,除了关注实验的各项注意事项外,高品质的产品同样至关重要,全式金生物提供Western Blot整体解决方案,产品性能优异,稳定性好,欢迎有需要的小伙伴联系我们。